
Allosteric Targets
The majority of existing drugs that target receptors bind in the same site as the endogenous agonist (the orthosteric site). Development of highly selective drugs in this space has been hampered by similarity of the orthosteric sites within groups of receptors with distinct function, tissue distribution, and associated disease areas. Recent developments have highlighted the fact that drugs can interact with alternative (allosteric) binding sites on the receptor molecule that are distinct from the orthosteric site.
Allosteric modulators offer several advantages over orthosteric ligands, including greater selectivity and saturability of their effect, but characterizing this mode of action has, until now, been challenging using existing technologies. BSI offers the ability to determine allosteric ligand affinity directly, as opposed to a functional readout of modulation of orthosteric potency and efficacy.
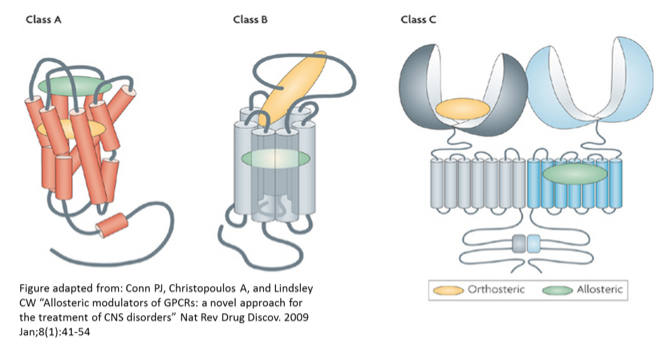
Different classes of G-protein-coupled receptors (GPCRs) have a common core structure of seven transmembrane (TM) helices, but have highly disparate orthosteric binding modes, reflected by their diversity of endogenous ligands. Class C GPCRs have far more spatial separation between orthosteric and allosteric binding sites than Class A and B GPCRs, and may have more than one allosteric pocket for small-molecule binding in the TM region.
The mechanism of action for an allosteric modulator can arise via conformational coupling between the orthosteric and allosteric binding sites (affinity modulation) or by altering the functional response of the receptor to orthosteric ligand binding (efficacy modulation). These mechanisms can have distinct pharmacological outcomes, thus determining which mechanism is dominant for a particular allosteric drug candidate through biophysical means can have significant value in the drug development process.
Back Scattering Interferometry (BSI) can illuminate these complexities through the characterization of distinct modes of allosterism, i.e. affinity- versus efficacy- driven mechanisms, through direct binding assays using intact membrane proteins.
Binding cooperativity in a class A GPCR
A key advantage of BSI is its applicability to mechanistic studies, being able to directly determine allosteric small molecule compound affinity for large, complex targets. Additionally, demonstration of conformational coupling between orthosteric and allosteric sites can bridge medicinal chemistry and pharmacology goals for refinement of drug profiles.
In the example below, the target, a Class A GPCR in a membrane preparation, was held constant at ~600 pM. An increase in allosteric ligand affinity (decrease in Kd) as orthosteric ligand concentration is increased identifies the titrated compound as a positive allosteric modulator (PAM). Further, demonstration of cooperativity between the allosteric and orthosteric binding sites identifies the mechanism of action for this type of allosteric compound as affinity-driven, which can have implications for medicinal chemistry development as well as refinement toward a particular pharmacological profile.
Increase in allosteric ligand affinity (decrease in Kd) as orthosteric ligand concentration is increased identifies the titrated compound as a positive allosteric modulator.
Lack of binding cooperativity in the class C GPCR, mGluR5
Membranes (both mGluR5 and non-expressing reference membranes) were diluted in PBS and mixed with serially-diluted PAM compound, incubated 2 hours at room temperature, and analyzed with the BSI device. Reference membrane signal was subtracted from target signal at each concentration point. Experiments were performed in the absence and presence of 30 mM glutamate.
The lack of affinity modulation for an allosteric modulator by the orthosteric ligand, glutamate, is reasonable given the large spatial separation between the binding sites. This result confirms the mechanism of action of this class of compounds as efficacy, rather than affinity, driven modulation of mGluR5.
Disparity in functional and radiologand binding assays for this system is highlighted below:
Functional data (left) that characterizes VU0405386 and CPPHA as potent PAMs is at odds with radioligand binding data (right) that suggests CPPHA is a weak binder. This can be explained by the presence of multiple allosteric binding sites on mGluR5, and reveals a shortcoming of radioligand binding assays, i.e., probe dependency.
BSI binding assays reveal that these two PAMs have similar affinities at their respective binding sites. This data is more closely aligned with the functional data than the radioligand binding assay that is hampered by probe-dependency at a single binding site.
Binding of low molecular weight GPCR ligands
BSI is also able to measure low affinity interactions due to its sensitivity to receptor conformational changes in free solution. Conventional radio-ligand binding assays are unsuitable for low affinity Kd’s due to downstream wash steps that allow dissociation of low affinity ligands before readout. The affinity of glutimate for the mGluR5 receptor is demonstrated below.
BSI enables binding quantification of very low molecular weight ligandsto large integral membrane proteins under conditions where other binding assay technologies fail. A Kd of 2.4 µM can be computed from the binding curve, which is comparable with data from other techniques.